Please wait to begin the exercises until the instructor has reviewed all instructions for each exercise.
Quick links to sections: Exercise 2 / Exercise 3 / Exercise 4 / Exercise 5
Completing all exercises (1 - 5) in lab today and transfering your 'work products' into the ELN is strongly recommended. If necessary, some of the analysis portion of Exercise 4 may be completed outside of class; however, for exercise 4, at a minimum complete your multiple sequence alignment (MSA) including the locating your AA 'variants' within your MSA, and understand how to complete the analysis on your own before leaving. [At least exercises 1-3, 4A, 4B, and 5 should be completed and in your ELN before leaving.] If you still have time left in lab, complete as much as possible before leaving - while help is still easily available.
'Work Products' for this lab - what should be completed and turned in (put in your ELN under appropriate headings).
You'll be provided a print out of your assigned DNA sequence. Using the genetic code (found near the end of this handout: DNA+AA info), fully translate the DNA sequence on your handout in all 3 frames showing all amino acids and stop codons predicted, and then mark the reading frame which contains an ORF (open reading frame). [Example]
Photograph your manual translation and post the legible image to the appropriate place in your ELN.
NCBI National Center for Biotechnology Information - access integrated biomedical/molecular databases, including Genbank.
Two other major sequence databases:
2. The European Nucleotide Archive (ENA) at the European Bioinformatics Institute (EBI), also associated with ExPASy (Expert Protein Analysis System) based in Switzerland.
These nucleotide databases are together part of the International Nucleotide Sequence Database Collaboration. From the main NCBI page, under Popular Resources (right side) click on "Nucleotide." Then search by entering your accession number (see your individual assignments) in the text box and click Search. The default format for the database entry is as a "Genbank Report" - you will see this indicated next Format:. Examine the GenBank report to see all the useful features and information. The nucleotide sequence itself is at the bottom of the page.
Now, change the selection next to Format: from GenBank to FASTA, by clicking on the link (red arrow & box in illustration below). This will retrieve the sequence in FASTA format (description), which is a minimal, simplified format recognized by most or all sequence analysis programs. This format is used both for nucleotide and amino acid sequences. Save this file.
![]()
ExPASy Translate tool - Allows 3 different output formats for your translation: "Verbose," "Compact," or including the nucleotide sequence.
1. Paste your nucleotide sequence (from Exercise 2) into the window. Be sure to paste in only DNA sequence.
2. Select an output format, and hit the TRANSLATE button. Starting with the default "Verbose" is specifically designed to help spot ORFs, highlighted in Red . This format also allows you to go on and select a specific ORF. Note that you can go back to the GenBank report for your nucleotide sequence and confirm you've identified the correct protein sequence.
3. Examine each of the 3 different ouput forms. Determine which reading frame is correct, and save that correct translation (e.g., copy & paste into a word document), in the "with nucleotide seq" format. Make sure to format neatly with a mono-spaced font and appropriate margins so the sequence is easily read.
Highlight the start and stop codon.
4. Using this tool, you can also go back and check your manual translation (from Exercise 1) using your DNA sequence, and the "with nucleotide seq" format. How well did you do the translation? Do you need to correct anything in your manual translation. If so, make the correction and upload the corrected version below your original picture from Exercise 1.
In this lab, you may imagine either of two possible future scenarios. 1)You work as a genetic counselor, and are analyzing information from a client or patient's DNA sequence, or 2) You have received information about your genes, or those of a friend or relative who has shared the information with you, and would like to evaluate the nature of variants found in the sequence of specific genes.
All of us have parts of our genome that are different from the most common sequence. (Did you know that the original / first human genome sequence from the Human Genome Sequencing Project was actually a composite made from a mix of many individuals' DNAs?)
The basic question: For any given AA variant, should I be concerned that the variant alters the function of the gene involved?
For this exercise, we will focus on small nucleotide changes that alter specific single amino acids in the protein coding region ('missense' alterations) of genes encoding mostly metabolic enzymes. Select one protein-of-interest from the list in this document: Human proteins associated with single amino acid change causing diseases or syndromes.
For each, there is a list of 'variants' or 'single nucleotide polymorphisms (SNPs)' or 'single nucleotide variants (SNVs)' found among human genomes. Each variant alters a single codon, and therefore changes a single AA in the protein from the most common version. Some of the AA variants are known to cause dysfunction in the protein ('pathogenic'). Others are neutral ('benign'), or, their effect on protein function is currently unknown.
You will analyze at least 6 missense variants found in your human protein of interest and evaluate the likely/possible effect of the AA variant (further instructions follow).
Parts 4A and 4B below will provide the information you need for analyzing your variants. Your analyses will be in Parts 4C and 4D.
Go to the NCBI Protein database and enter the accession number.
Get the protein sequence in FASTA format. You will see a link for that on the page. Copy this into a text file.
Now copy and paste your POI sequence (don't use the accession number) into ExPASy protein parameters tool to get a nicely numbered version that will make it easier to find specific AAs (i.e., your sequence variants). It will appear at the top of the ProtParam result. Example of a numbered sequence shown below:

You can ignore all the other useful info below. Copy the top part into a file and then mark the locations of all your variants in this numbered version (e.g., highlight or bold).
You will need this information to go with your multiple sequence alignment.
[Further instructions: How to find a variant location.]
Making alignments of related protein sequences is an important bioinformatic technique for a variety of purposes. Alignments show what portions of a molecule are shared between two or more molecules.
Parts of a given protein that are highly conserved (that is, unchanged over evolutionary time) in many distantly-related organisms are likely to be important functional regions. In other words, that portion of the protein cannot be altered by mutations without destroying or reducing the protein's function. Organisms with such mutations are selected against - out-competed for reproductive success - so the changes are lost. On the other hand, regions of the protein (or specific amino acids) that can change without disrupting protein function will evolve - randomly change over time - and be different in distantly-related organisms.
B1. Download seven more protein sequences to align (total 8, including your human POI)
In order to find conservation related to function, it will be best to acquire orthologs from a broad range of phylogenetic distances, such as those in the list of different species below: (Example FASTA file + species list). Ideally, choose sequences all from different animal phyla. You already have one vertebrate (human), so include no more than one other chordate (ideally, a non-vertebrate chordate). See here for an animal phylogeny showing some major phyla, plus names of some animals with fully sequenced genomes. (If you're having a problem finding sequences from 7 different phyla, then use more than one in a given phylum, but no more than 2 per phylum.)
a. Go to your gene/protein page by entering 'Human Protein-Name*' into the search line at the NCBI Protein database (and searching).
* the abbreviation for the protein in parenthesis in the list of proteins and variants, e.g., 'Human AMT'
This should result in a screen that looks like this (for a protein called WNT1):
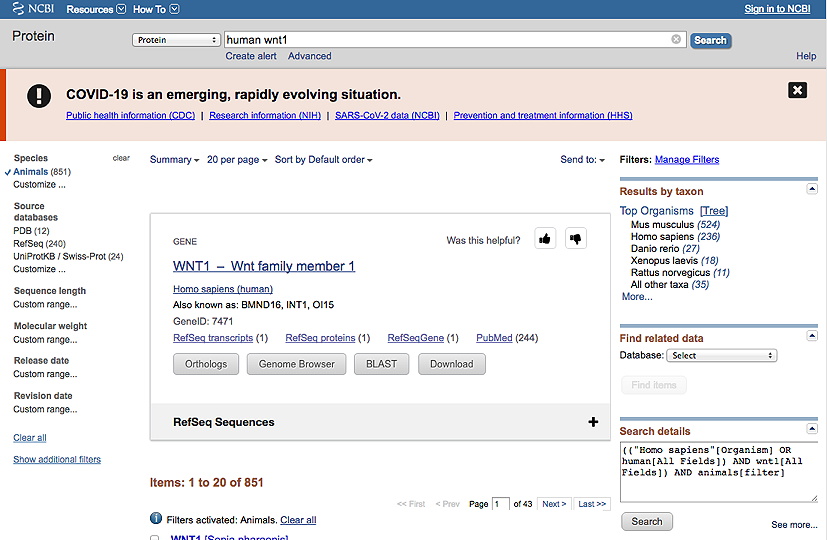
b. Then, under Species in the lefthand column, click Animals.
c. Then, in the righthand column below Results by taxon, click on Tree. The example shown below indicates there are sequences available from 6 different phyla (chordates, arthropods, flatworms, molluscs, nematodes and cnidarians). [So for this example, you would have to take more than one sequence from a given phylum to get up to 8 sequences total.]

d. Click on the number next to the group name to get a list of sequences from that group (or, if there's only one, it will take you directly to that sequence page). Be sure to collect protein sequences of a very similar length to that of the human protein. True orthologs are typically within 5-20 AAs of the same length. Also avoid sequences with a description like 'Low quality' or 'partial' or 'Protein-name-like.'
On the other hand, 'hypothetical' is OK - just still look for a similar length of protein.
d. Acquire all the protein sequences in FASTA format.
B2. Paste all sequences in FASTA format into a single file
In one method of performing a multiple sequence alignment, we must first create a plain text file with all the sequences we wish to align in the FASTA format. Open a word processor window on your computer (and stagger the two windows on the screen so you can easily go back and forth between them). Then paste each sequence into a single text document, with a blank line in between each. [Again, see an example FASTA format file.]
Put the human sequence at the top, since this is the one you'll be comparing to the others.
B3. Create a new file and rename the sequences
Rename each sequence in your file with the genus & species name initials, underscore, and an abbreviated protein name. (You may observe in GenBank files for your protein that there is a commonly used short name, for example.) Make sure there are no spaces in the name. Keep track of the identity of your all your proteins - don't discard the accession number information and full description, for example - you may need it later.
Example: rename the full FASTA description
>gi|132814447|ref|NM_001082971.1| Homo sapiens dopa decarboxylase (aromatic L-amino acid decarboxylase) (DDC), transcript variant 1, mRNA
as:
>Hsa_DDC
- In this example, a single capital letter is used for the genus, followed by the first two letters of the species name: Homo sapiens becomes 'Hsa'.
Create a separate file with the species abbreviation information like this example that includes the abbreviation, full species name, and common name.
B4. Run the alignment with Clustal Omega.
The program we will use for alignment is Clustal Omega and is found at the European Bioinformatics Institute (EBI) website. There are other multiple sequence aligmnent programs available, including on the NCBI website.
Under 'STEP 1,' paste the collected sequences into the large window.
Under 'STEP 2,' click on 'More options...' to change this parameter:
ORDER (last among all the options): change default 'aligned' to 'input' - this keep your human POI sequence at the top of the MSA.
Part of an example alignment with this format (using the GCH1 proteins file) is shown below:

It's not required, but you might like seeing and saving a colorful version - click 'Show Colors' (example is shown in section 4D below). It does provide some additional information.
If you get a poor alignment, consider removing (and replacing if below 8 sequences) a protein sequence that may be disrupting the alignment. Occasionally a protein you've chosen is quite different from all the others.
Files you can use for demonstration purposes: Example FASTA files
i. What is the type of AA (original) vs. the variant? There are a variety of ways amino acids can be classified - see the AA info handout.
ii. What is the BLOSUM score (using the BLOSUM62 substitution matrix*) for the change?
[* Where did the BLOSUM62 alignment score matrix come from? ]
iii. From this information, do you think this AA change is a minimal, moderate, or radical change?
Now, supplement your analysis based on AA type change with critical information derived from your MSA - the AA's specific location in the protein. How conserved is the AA is at this location?
Examine your MSA carefully - at the bottom of each stack of AA's at a given location is an indicator of conservation level (see below): either no clear conservation (no marker), a period, colon, or asterisk. This indicates what level of change the protein can tolerate at this site. For example, even if a change is 'mild' wherein a very similar AA is substituted (e.g., an Isoleucine for a Leucine), if the Leucine is 100% conserved, this might still be a radical (pathogenic) change.
On the other hand, at some locations any AA can be tolerated, so even a radical AA type change is likely to be benign.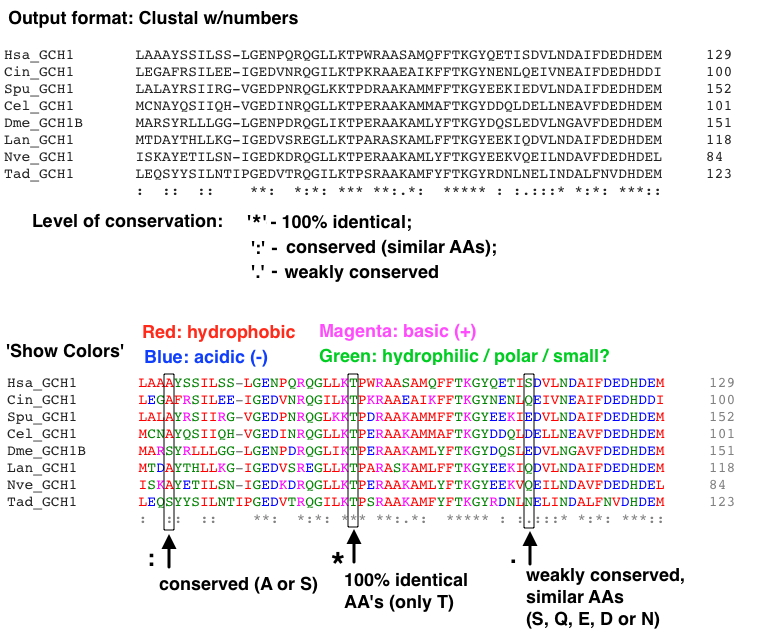
1. Find the crystal structure for your protein of interest. First, get the PDB ID - like an accession number - for your protein can be found in the proteins & variants file you used previously. These identifiers are made of 4 capital letters and numbers. Some examples: 2GK1, 1HD.
2. Go to NCBI - Structure and search with the PDB ID. You should reach a page that looks like below (here a crystal structure for Human GTP Cyclohydrolase I - 1FB1). To see the structure dynamically, click (red arrow) on 'full-featured 3D viewer.' This will take you to iCn3D - "I see in 3-D" -NCBI WebGL-based Structure Viewer page, and load your structure. This may take a few seconds.
![]()
Once your structure is loaded, you'll be able to find the locations of your variants. [Check whether the AA numbering is different from that of the full-length protein.]
a. Under the Analysis menu, select 'View Sequences & Annotations,' and then select the 'Details' tab to see the AA sequence alongside the structure.
b. Select an amino acid or range of amino acids (click and drag across the AA) in the primary sequence to highlight the location of a variant in the structure. (The highlight is a small yellow region. You may need to rotate the protein to see it clearly.)
Does the location of the mutation provide any insight into why it might disrupt the function of the protein? AA's at the surface or in extended structures are somewhat less likely to disrupt function when altered. AA's deep within the protein or tightly packed are more likely to affect function if changed. If the protein is multimeric, AA's at the interface between subunits may be critical for (e.g.) dimerization or tetramerization. There may also be crystal structures of your protein interacting with a binding partner.
c. Take a screenshot of the 3D protein structure you viewed in iCn3D for each amino acid variant location, with the AA selected to turn in. Include the part of the right side window with the AA sequence showing which AA was selected.
An example is shown below. In this case, the AA (R88) was selected in two copies in the sequence window to show up on 2 of the 3D protein chains of the tetrameric crystal structure. The program highlights the location in yellow. If you move your cursor over the highlighted location, you can also get the amino acid information to appear (here 'ARG88').
NOTE: In many cases, the AA numbering of the full-length protein does not match that found in the crystal structure.* If this is the case for your protein, be sure to add something at the beginning of this section (before your screenshots) that indicates how the numbering has changed: i.e., for each AA position, indicate the numbering in the crystal structure.
For example, "The crystal structure protein is missing the first 31 AAs of the full-length protein. Therefore, R233 (full-length) = R202 (233-31 = 202, crystal structure); L236 = L205; T276 = T245; I440 = I309; etc."
[* Why is this? Often, a truncated version of a protein forms a better crystal than the full-length version, or the full-length protein won't crystallize at all! These are the vagaries of the 'art' of protein crystallization.]

Example of Completed Exercise 4