In your first bioinformatics lab during week 1, you made your own analyses of human protein sequence variants for the gene you chose. First, go back and review that exercise (Lab 1, exercise 4) and your analyses as found in that section of your ELN. Retrieve the list of variants that you analyzed, plus the accession number for your protein of interest.
Recall the scenario:
Either now, or in the very near future, most or all of us will be acquiring complete sequences of our genomes, either associated with a particular clinical presentation (e.g., cancer or other syndrome, or developmental abnormality of a child), or as a routine part of one's preventative / prognostic medical workup.
In this lab, you may imagine either of two possible future scenarios. 1) You work as a genetic counselor, and are analyzing information from a client or patient's DNA sequencing, or 2) You have received information about your genes, or those of a friend or relative who has shared the information with you, and would like to evaluate the nature of variants found in the sequence of specific genes. All of us have some parts of our genome that are different from the most common sequence.
The basic question: For any given variant, should I be concerned that the variant alters the function of the gene involved?
In this lab, let's take advantage of expert systems that can provide additional analyses, and see what assessments have already been made of your variants.
Links to all the websites you'll be using for this lab (also found within the instructions below):
Poly-Phen-2 |
SIFT |
UniProt |
NCBI ClinVar
Our example/demonstration sequence (used for all the examples shown below): Human Wnt1
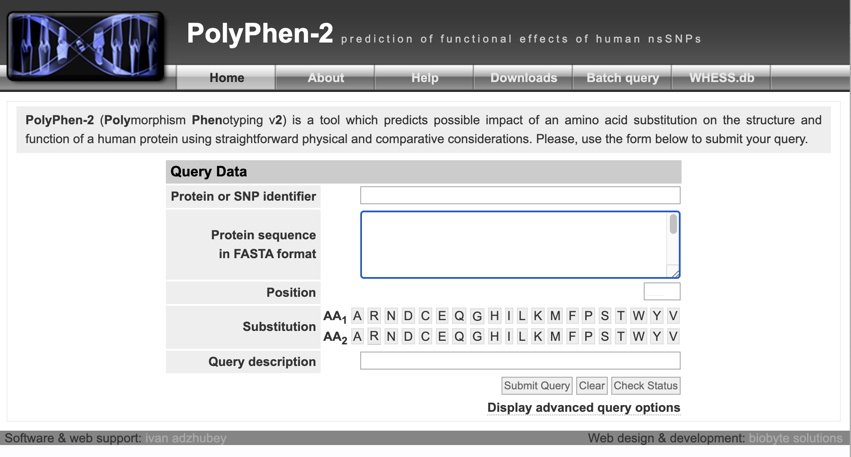
Poly-Phen-2 - click to access the window shown below:
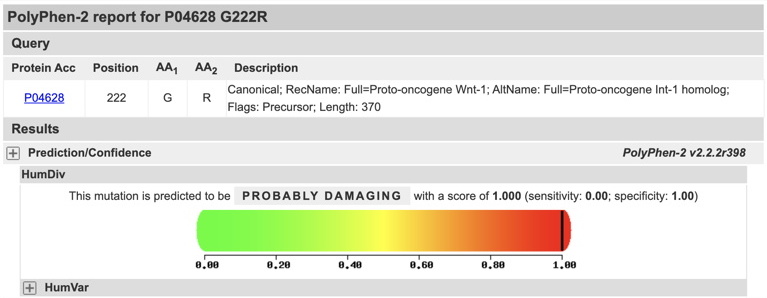
Enter the appropriate information for each of your variants (as illustrated below for the Wnt1 protein and the variant G222R):
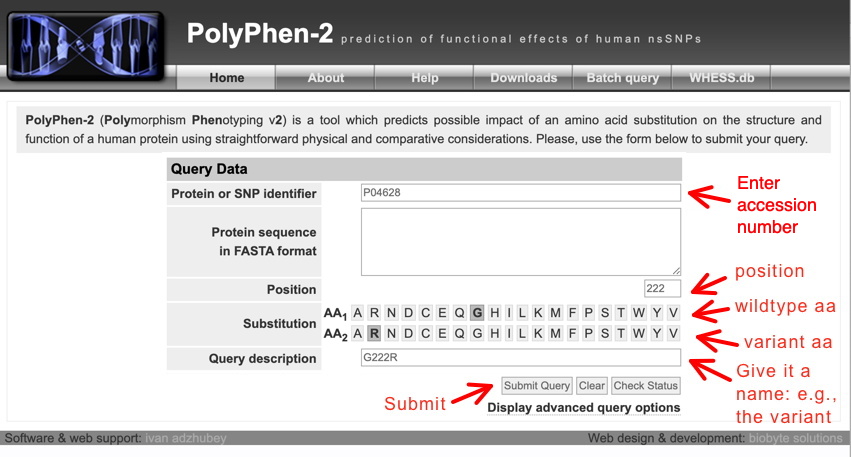
This will yield the following (click View to see the report); if View is not there yet, click Refresh after 5-10 sec until it shows up. Note you can go back to the PolyPhen-2 window and submit the rest of your variants - each job will still be available (accessible with View).
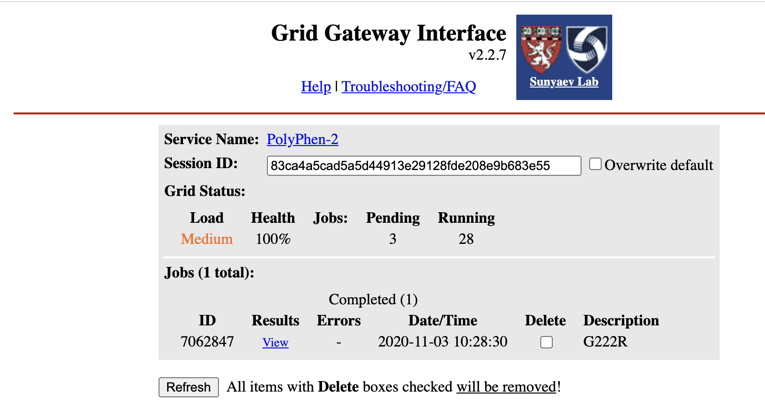
Example of PolyPhen-2 report for G222R variant (below):
Click on + Multiple sequence alignment to see the one created by PolyPhen-2 (below). If you wish, you can also examine the 3D visualization (like what you did with iCn3D in week 1). Note that much of this MSA is not as useful as the one you prepared in week 1 because the alignment includes a lot of closely related proteins with high sequence identity - AAs are conserved because of recent species divergence rather than because of a functional requirement.

Take a screenshot of the PolyPhen-2 report (Prediction/Confidence) for each of your variants (the MSA and 3D visualization is not required, although you may find them interesting to examine). An example of an appropriate screenshot is shown below.
Check out a second predictor of protein sequence function. How does SIFT work?
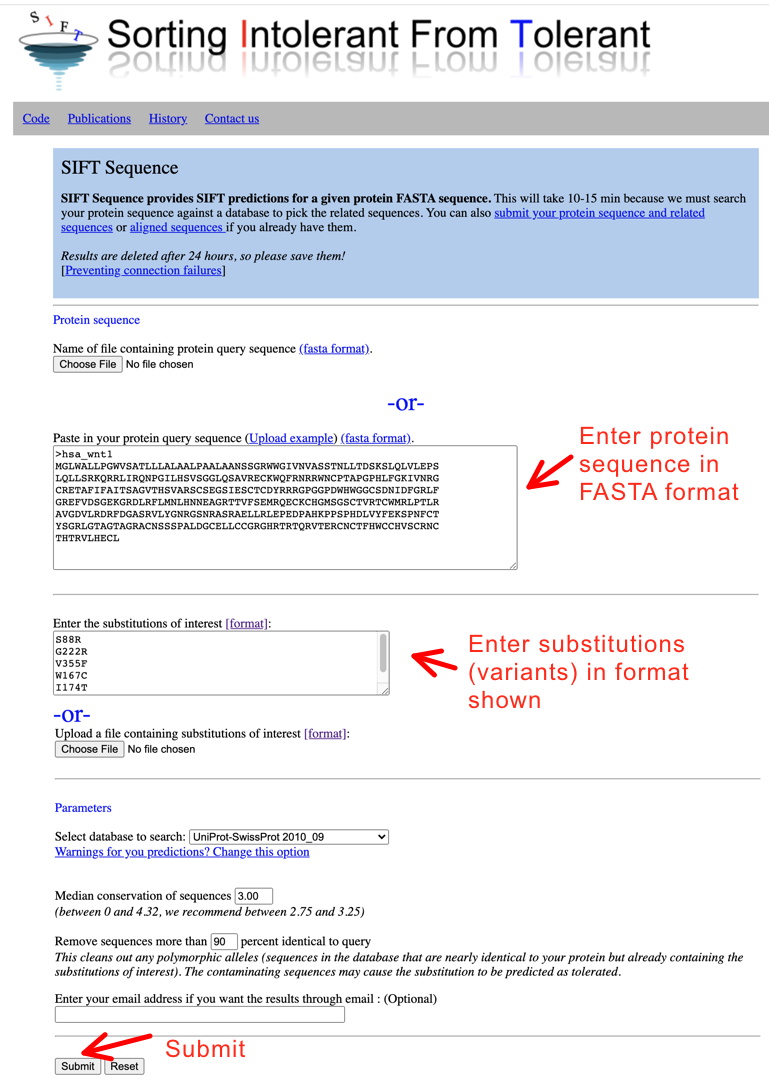
Using SIFT: Go to the page, paste in your protein sequence of interest in FASTA format, enter the variants of interest, and submit (illustrated below).
Conveniently, with this program, you can enter all your variants at once.
The initial report page will look like this:

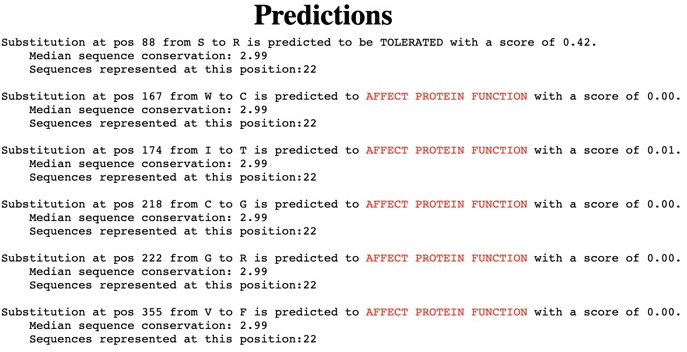
First, click on the link for the 'Predictions of substitutions entered' (lower arrow) - our example for Wnt1 with the entered substitutions is shown below. Screenshot this set of predictions for your variants. (SIFT output for a single protein analysis - interpreting these results - see especially 3.)
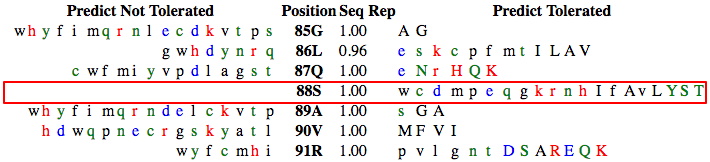
Next, check out the cool predictions for all possible substitutions at every position in your protein (link indicated by top arrow above). Positions in the protein are represented top to bottom. The AAs on the right are predicted to be tolerated substitutions at that position (capital letters are found at that position in the MSA that SIFT made and analyzed); the AAs on the left are predicted NOT to be tolerated substitutions. See below the 'Predictions for positions 1 through 100' for the meanings of the letters - colors (green, red, blue), etc.
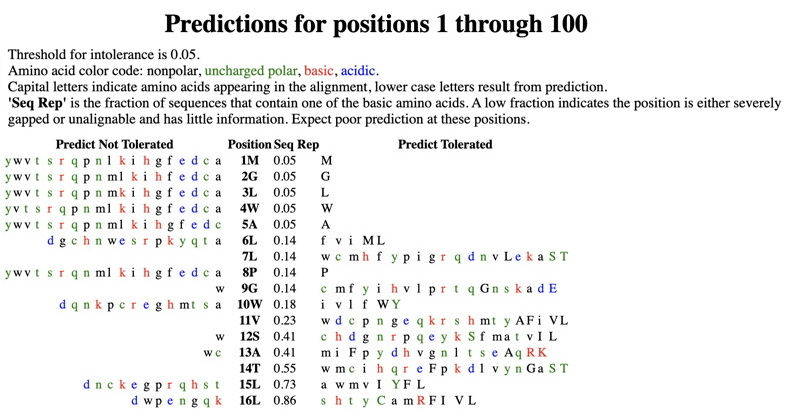
Find the region surrounding each of your variants. Screenshot this region including your variant and 3-4 AAs above and below. Highlight your amino acid location. An example is shown below for position 88 (S88R) in the Wnt1 protein. For this example, it is clear that substitutions at this site are likely to be tolerated -- every AA at that site is predicted to be tolerated. These predictions show how readily any substitution is likely to be accepted at your specific variant locations.
First, look for the UniProt page using your accession number -- that may take you directly to the correct page. If not, try going back to the full NCBI protein entry [NCBI Protein database] to find either a link to the the UniProt entry, or to otherwise locate the UniProt number. You can also try to find your protein's UniProt page using the protein name as a keyword.
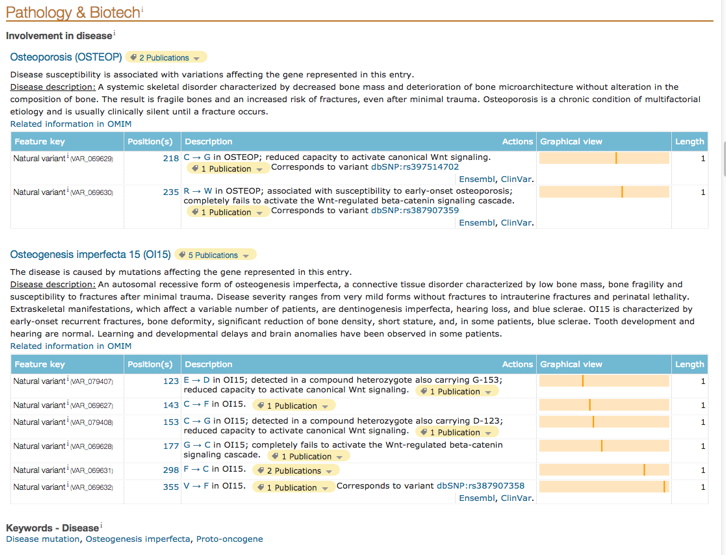
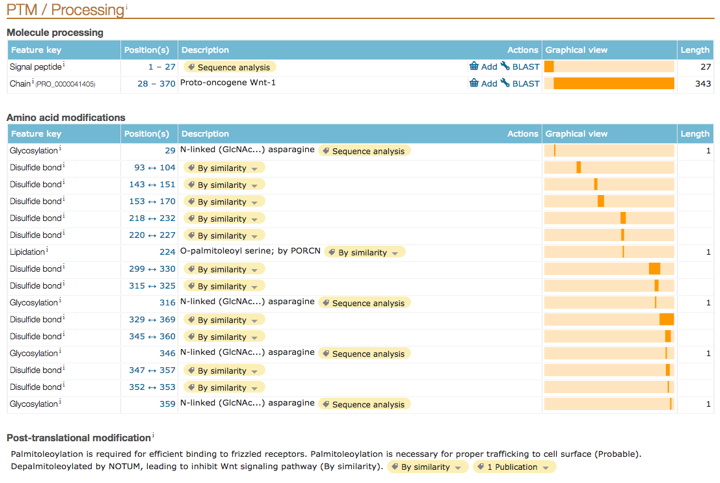
Here are just two useful entries - more variants (and pathology), and protein structure-function information. Record any relevant information for your protein variants. If there is none, indicate that result from your UniProt analysis.
Question: According to the UniProt information (especially in the 'Pathology & Biotech' section), are any of your specific variants known to cause a specific abnormality / disease / condition / syndrome in humans?
In the 'Amino acid modifications' section ('PTM/Processing' section), do any of those locations match up with your variants? If so, note this as an indication of a reason why changing the AA at that location might be detrimental.
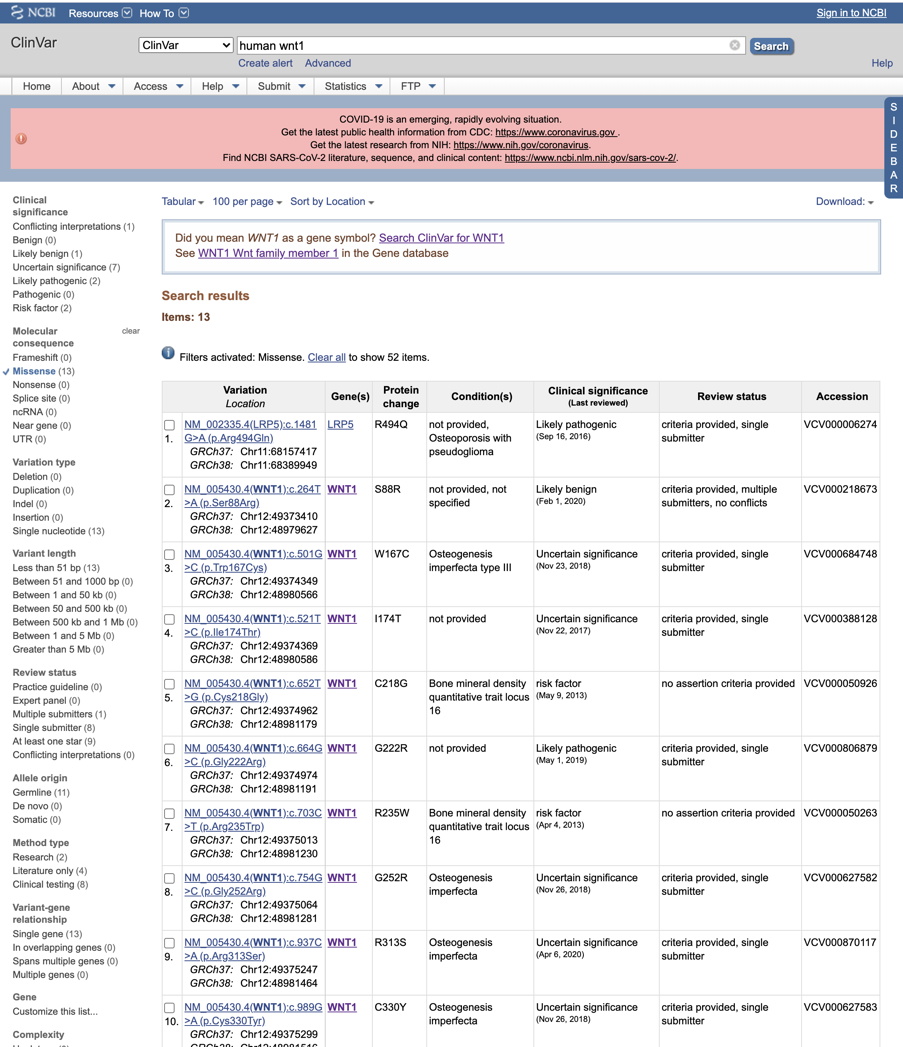
Note that sometimes other genes come up in the list (not your gene of interest) because of keywords. Below, at the top of the list we see a variant in the gene LRP5 which is a receptor for the Wnt ligand. Record the 'Clincal significance' information for each of your variants, or screenshot each line containing one of your variants. Note that the 'Review status' information is helpful for evaluating the reliability of the analysis.
Discussion - compare your conclusions from lab 1, exercise 4 with those from each new analysis - how well does your prediction hold up? Also compare all of the new analyses - how consistent are they? From the UniProt and ClinVar analyses, you can comment on actual / known protein dysfunction (leading to a genetic condition) caused by a variant, not just a prediction.