You should complete analyses of Exercises 1 - 3 and 5 - 7 in lab today and transfer your 'work products' into ELN.
It may be possible to complete the entire exercise during lab - this is recommended.
If necessary, Exercise 4 may be completed outside of class; however, you must get it started so that you can ask questions.
You must at a minimum complete your multiple sequence alignment (MSA) and add to your ELN.
Using the genetic code handout, translate the DNA sequence on your handout in 3 frames, and determine the reading frame which contains an ORF (open reading frame). Turn in your manual translation today before you leave (with your name, of course). All other materials should be added to your ELN.
ExPASy Translate tool - Allows 3 different output formats for your translation: "Verbose," "Compact," or including the nucleotide sequence.
1. Paste your nucleotide sequence (from Exercise 2) into the window. Be sure to paste in only DNA sequence.
2. Select an output format, and hit the TRANSLATE button. Starting with the default "Verbose" is specifically designed to help spot ORFs, highlighted in Red . This format also allows you to go on and select a specific ORF. Note that you can go back to the GenBank report for your nucleotide sequence and confirm you've identified the correct protein sequence.
3. Examine each of the 3 different ouput forms. Determine which reading frame is correct, and save that correct translation (e.g., copy & paste into a word document), in the "with nucleotide seq" format. Make sure to format neatly with a mono-spaced font and appropriate margins so the sequence is easily read.
Highlight the start and stop codon.
4. Using this tool, you can also go back and check your manual translation (from Exercise 1) using your DNA sequence, and the "with nucleotide seq" format. How well did you do the translation?
Exercise 4 - Multiple sequence alignment of protein sequences, using MSA to predict effects of single amino acid changes on protein function
Making alignments of related protein sequences is an important technique for a variety of purposes. Alignments show what portions of a molecule are shared between two or more molecules. Alignments are the starting point for determining the relatedness of molecules, and by extension, the organisms from which they come.
Parts of a given protein that are highly conserved (that is, unchanged over evolutionary time) in many distantly-related organisms are likely to be important functional regions. In other words, that portion of the protein cannot be altered by mutations in the gene encoding it without destroying or reducing the protein's function. Such mutants are selected against and do not survive. Regions of the protein that can change without disrupting protein function will evolve over time and be different in distantly-related organisms.
[See this handout for amino acid properties info: DNA+AA info.]
Select one protein-of-interest from the list in the document accessed below:
Human proteins associated with single amino acid change causing diseases or syndromes -- For each, there is a list of 'variants' or 'single nucleotide polymorphisms (SNPs)' found among human genomes. Some of these are known to cause dysfunction in the protein. Others are neutral, or how they alter protein function is unknown.
For your protein-of-interest, select 5 variants (single amino acid changes) to analyze. You will do this in part by making an MSA to analyze sequence conservation:
1. Download protein sequences to align
Using your newly acquired sequence retrieval skills, first acquire your protein of interest from the NCBI Protein database using the accession number provided.
This will insure you have the proper form. For many proteins, there are several versions of different lengths arising from alternative RNA splicing. This one should have the same numbering system for the amino acids as found in the list of variants. Click on the link to get the FASTA format (left red arrow below), and copy this into a plain text file.

Then use Protein BLAST (right red arrow above) to find and save the sequences of at least 5 related proteins in FASTA format (more is probably better).
First, acquire the human protein for your analysis using the provided accession number. This will take you to a blastp page, and automatically insert the accession number in the window for a blastp search (see image below).
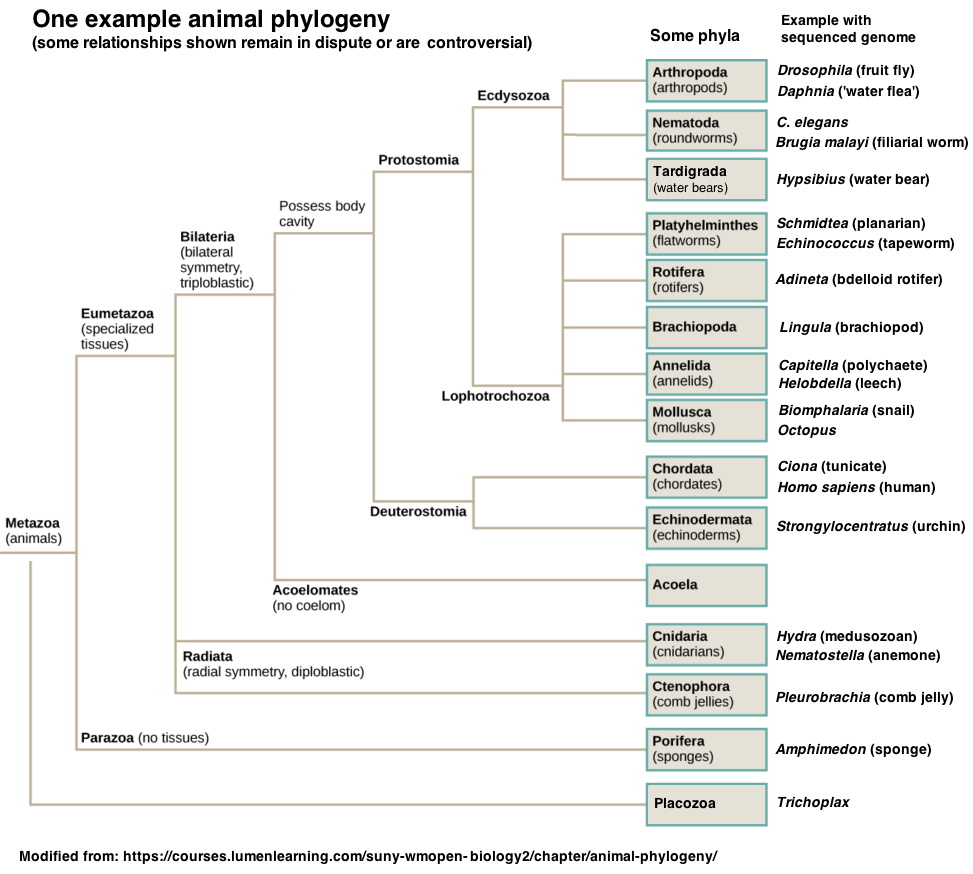
In order to find conservation related to function, get proteins sequences from a broad range of phylogenetic distances, such as those in the list of different species below: (Example FASTA file + species list). Choose sequences all from different animal phyla. See here for an animal phylogeny showing some major phyla, plus names of animals with fully sequenced genomes. You can select specific phyla or a species within a phylum by typing the name into the optional Organism box (red arrow below). Choices will appear as you begin to type.
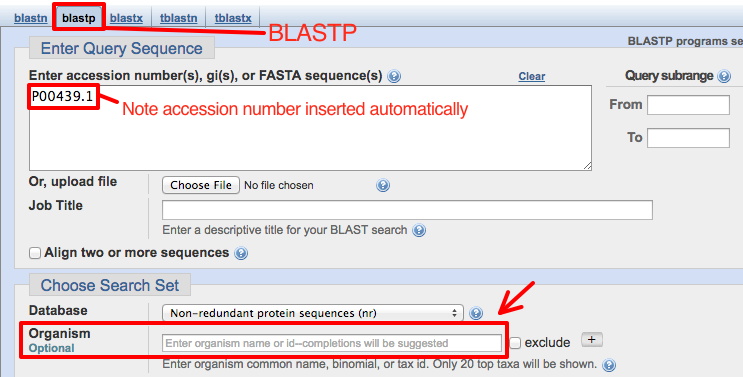
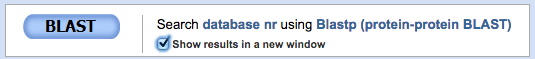
Since you will want to run this BLASTP several times searching different phyla, select Show results in a new window before clicking the BLAST button (see below). Then you can go back to this page to perform a new search by replacing the phylum name.
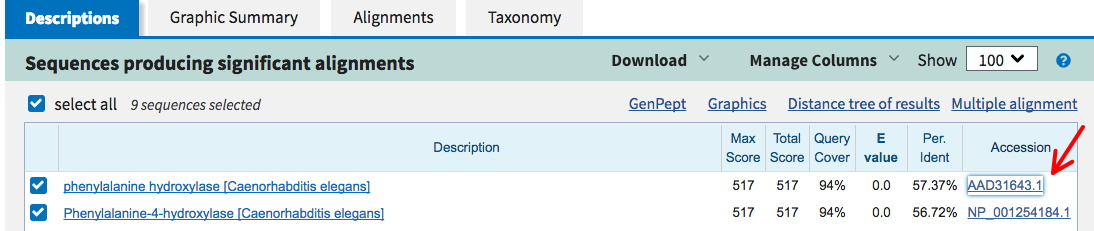
From the BLAST result page, click on an Accession number to go to the GenBank report for that protein (in another species). Click on FASTA there to get that form of the protein sequence. [To get this result, the species name Caenorhabditis elegans was entered into the Organism box (phylum, Nematode).]
2. Paste all sequences in FASTA format into a single file
In one method of performing a multiple sequence alignment, we must first create a plain text file with all the sequences we wish to align in the FASTA format.
Open a word processor window on your computer (and stagger the two windows on the screen so you can easily go back and forth between them). Then paste each sequence
into a single text document, with a blank line in between each. [Again, see an example FASTA format file.]
Put the human sequence at the top, since this is the one you'll be comparing to the others.
3. Rename the sequences
For convenience (given the program we are using), in your sequence collection file, rename each sequence with the species name initials, underscore, and short name. (You may observe in GenBank files for your protein that there is a commonly used short name, for example.) Make sure there are no spaces in the name.
Example: rename the full FASTA description
>gi|132814447|ref|NM_001082971.1| Homo sapiens dopa decarboxylase (aromatic L-amino acid decarboxylase) (DDC), transcript variant 1, mRNA
As:
>Hsa_DDC
4. Run the alignment with Clustal Omega.
The program we will use for alignment is Clustal Omega and is found at the EBI website.
There are other multiple sequence aligmnent programs available, including on the NCBI website.
Either paste the collected sequences into the large window, or upload the saved file (note that for uploading the file MUST be in a plain text/text only format).
Although we will mostly use the program default settings, change these two output parameters:
OUTPUT FORMAT - you can try a couple different formats to see the possibilities - but this one will be most useful:
'Clustal w/numbers' - shows the amount of conservation below the alignment, with AA coordinates at the ends.
[Note, once the alignment is displayed there is a lovely option to make the AAs color coded - 'Show Colors.']
ORDER (Click the 'More Options...' button): change this selection (lower right) from the default 'aligned' to 'input' - this will retain the order of sequences in your input file - i.e., keep the human sequence at the top.
[Note that the order in which the sequences are input (that is, the order you have them in your FASTA file) can affect the alignment.]
Part of an example alignment with this format (using the GCH1 proteins file) is shown below:
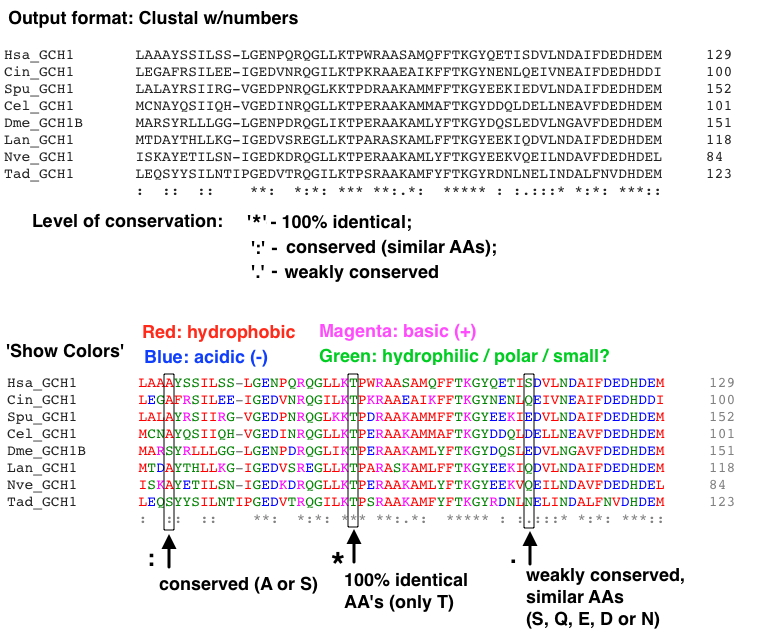
If you get a poor alignment, consider removing (and replacing if below 5 sequences) a protein sequence that may be disrupting the alignment. Occasionally a protein you've chosen is quite different from all the others.
Files you can use for demonstration purposes: Example FASTA files
Copy and paste your human protein-of-interest sequence (don't use the accession number) into ExPASy protein parameters tool to get a nicely numbered version that will make it easier to find specific AAs (i.e., your sequence variants). It will appear at the top of the result. You can ignore all the other useful info below. Copy it into a file for use when you do your variant analysis. Example of a numbered sequence shown below:

What should be in your completed protein sequence variant analysis with MSA:
- MSA with at least 5 protein sequences from 5 different phyla, with the human protein-of-interest at the top of alignment. Highlight the AAs in the human protein the variants of which you analyzed.
Each protein should have a 3 letter organism identifier and short name. Include a list identifying the organisms below the MSA, with species name and common descriptor.
- Analyze at least 5 missense variants found in your human protein of interest.
Evaluate the likely/possible effect of the AA variant based on:
a. Change in the type of AA, based purely on AA properties and empirical substitution frequencies.
b. AA location in the protein - how conserved the AA is at this location, etc. As noted above, mark the location of the variant AAs in the human protein in the MSA.
c. (optional) - If you examine your protein in Exercise 5, you may be able to see where the AA is located in the 3D structure (see below).
Exercise 5 - Access the Protein Structure Database, View Structures
1. Go to NCBI - Structure
2. Search with one of the following accession numbers:
- 1TAQ - Taq DNA Polymerase I
(enzyme for routine PCR)
- 1JZ7 - E. coli Beta-galactosidase (from lacZ gene) in Complex With Galactose
Or, try your protein of interest from Exercise 4. You can search for it in the NCBI Structure database. [Note: some of these proteins may not have crystal structures.] AA's at the surface or in extended structures are somewhat less likely to disrupt function when altered. AA's deep within the protein or tightly packed are more likely to affect function if changed. If the protein is multimeric, AA's at the interface between subunits may be critical for (e.g.) dimerization or tetramerization.
3. Click on the entry, which will take you to a MMDB Structure Summary
4. View structures using iCn3D - "I see in 3-D" -NCBI WebGL-based Structure Viewer page
When you're ready to use it, click on "OPEN iCn3D" (see image below).

Then enter a PDB file number by clicking FILE, and 'Retrieve by ID' - PBD ID, and wait for the image to load. (Or if you Then you can view it in a variety of ways.
a. Under the Windows menu, select 'View Sequences & Annotations,' and then select the 'Details' tab to see the AA sequence alongside the structure.
b. Select an amino acid or range of amino acids in the primary sequence to highlight the location in the structure.
Use this to find the location of mutations in your protein of interest from the multiple sequence alignments.
Does the location of the mutation provide any insight into why it might disrupt the function of the protein?
c. Take a screenshot of the 3D protein structure you viewed in iCn3D (with an amino acid selected) to turn in. Include a part of the window with the AA sequence showing which AA was selected.
Exercise 6 - Use a 'Genefinder' to perform an ab initio gene prediction on genomic DNA
Using nematode genomic sequence ( Oscheius myriophila, a relative of the better-known C. elegans) is convenient because gene density is relatively high
- that is, there are typically several genes in a relatively small sequence.
Directory of O. myriophila genomic sequences. Find your assigned sequence genomic sequence and save it to your computer (as a plain text file).
Wed section / Thurs section.
FGENESH - a gene prediction program.
[Note: we may have a limited number of free uses from USD's internet domain - use it ONCE today and get your info.]
1. Given the size of sequence, it's probably best to use the 'Choose File' button to select the file from your computer.
[Or, paste your assigned genomic sequence into the sequence window.]
2. Select 'Caenorhabditis elegans' for Organism (start typing in the window and it will show up).
3. Click [Show advanced options] to see what's available, and:
unselect 'print mRNA sequences for predicted genes' since we won't be using it.
4. Click the "Search" button.
5. How many genes are predicted in your genomic sequence? Are they large or small? Do they have few or many exons?
Calculate a measure of the (predicted) gene density: Divide the total sequence length by the number of predicted genes - this tells you that, on average, a gene is found every N basepairs.
Compare your calculations to others at your bench.
Guide to interpreting the FGENESH results
6. Save the resulting file to use in Exercise 7 below (and for a brief report on the results). You will use the predicted protein sequences in exercise 7B.
The PDF version creates a nice graphical view of each predicted gene plus all the sequences (and is easier to save).
Optional: Compare the genes found with FGENESH with another prediction program:
GeneMark.hmm - how similar or different are the results? [As with FGENESH, choose C. elegans.]
It may also be interesting see how the predictions differ if (using FGENESH again) you select 'Pristionchus pacificus' for Organism instead - this is the next closest nematode relative in the list.
The predictions are likely to be similar but not identical.
Exercise 7 - Use BLAST to assess your FGENESH ab initio gene predictions - e.g. how many of your 'predicted genes' are likely real genes?
A. Search for genes in your genomic sequence by homology using BLASTX - can strengthen genefinder predictions from above, but also may find genes NOT predicted by the genefinder.
1. From the NCBI BLAST Server again, click on blastx.
2. Choose your assigned genomic sequence to upload. Enter 'animals' in the Organism box (as shown below). We also need to change some default parameters - see the image below for the changes - they are highlighted in yellow, as they will be
when you make the changes. First, change the database, and then click on Algorithm Parameters to make the other changes.
Change the Max target sequences to 50, and Expected threshold to 1 (as shown below).
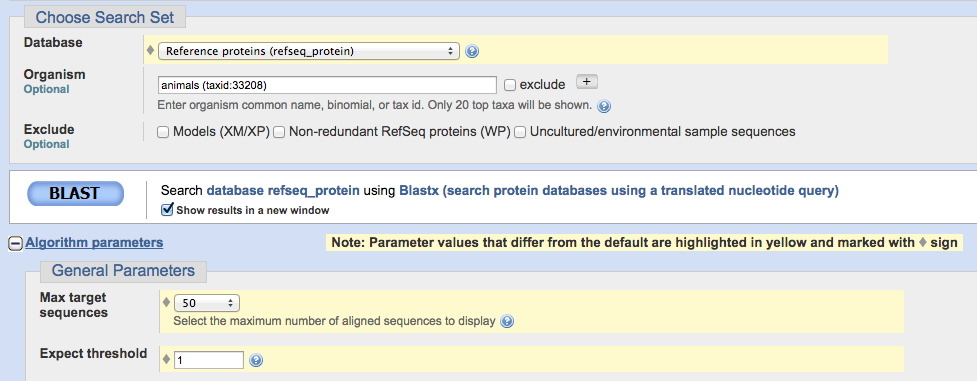
After parameters are changed, click the BLAST button, and examine the results.
(Note that this search may take a few minutes to be completed - while you're waiting for that window, you could begin part 7B below.)
Note the locations of homologies detected relative to proteins predicted by FGENESH - how well do they match up? Did FGENESH miss predictions of some regions detected by BLASTX?
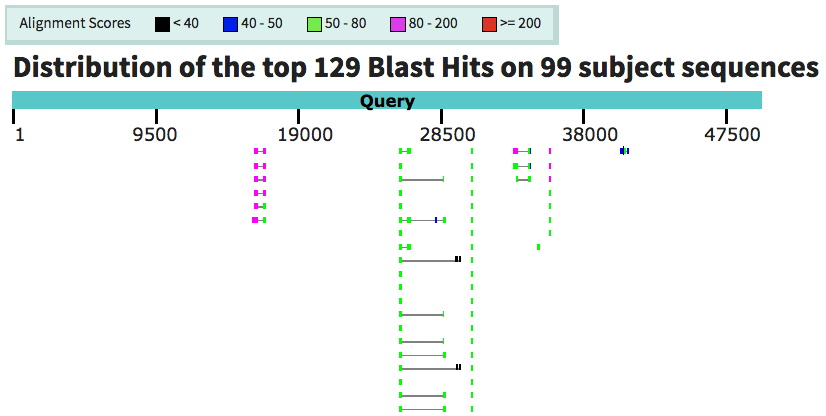
Below: example of the graphic view showing matches in your submitted sequence. The boxes joined by lines are part of the same match, and likely represent exons of a given gene.
(Note the color indicates the strength of the match, as shown in the key above.) This sequence shows 4 multi-exon likely genes, and 2-3 individual sequences matches
- 6-7 possible genes identified by similarity to genes from other organisms. For this sequence, this was fewer than predicted by FGENESH, but some of those shown here are the
same as FGENESH-predicted genes.
Optional: A more computation-intensive approach that can find matches with previously unidentified (never predicted) proteins would use tblastx.
Are there any regions detected with TBLASTX that were not detected with BLASTX? Make a note of these regions, if found.
These methods compare everything (the entire genomic sequence) at once, and can be a bit overwhelming initially.
The method below (7B) searches for matches to your predicted proteins one at a time.
B. Determine whether individual proteins predicted by FGENESH have matches in the protein database.
1. Go to the NCBI BLAST Server
2. Click on Protein BLAST (blastp).
3. Paste each predicted amino acid sequence from your FGENESH analysis indvidually into the large window under "Enter Query Sequence."
All the other parameters can be left at their default settings.
4. Select the "Show results in a new window" checkbox, then click the BLAST button.
(Record which of your genes appear to encode proteins known from other organisms. Consider what 'E-values' represent genuine matches with related proteins vs. random matches.)
Optional: Note Conserved Domains found in your search. (This information will come up immediately after initiating the search in the 'Format' window.) This is a strong indication that your sequence encodes a real protein.
Reminder: 'Work Products' for this lab - what should be completed and turned in (put in your ELN) - includes what is needed from Exercises 6 & 7 (mostly a simple table).
© 2020 Curtis Loer, Dept of Biology, USD. All rights reserved. Simple, hand-coded pages.
{kind=link}