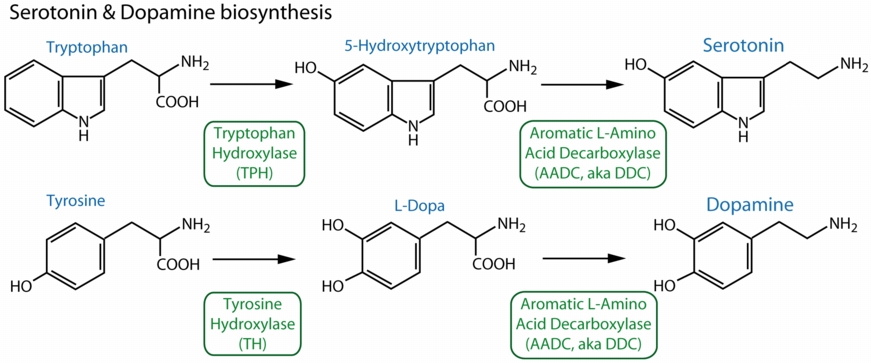
Figure illustrating function of aromatic L-amino acid decarboxylase (AADC) aka dopa decarboxylase (DDC) proteins in animals.
Making alignments of related protein sequences is an important technique for a variety of purposes. Alignments show what portions of a molecule are shared between two or more molecules. Alignments are the starting point for determining the relatedness of molecules, and by extension the organisms from which they come.
Parts of a given protein that are highly conserved (that is, unchanged over evolutionary time) in many distantly related organisms are likely to be important functional regions. In other words, that portion of the protein cannot be altered by mutations in the gene encoding it without destroying or reducing the protein's function. Such mutants are selected against and do not survive. Regions of the protein that can change without disrupting protein function will evolve over time and be different in distantly related organisms.
1. Download sequences to align
Using your newly acquired sequence retrieval skills, find and save your protein sequences in FASTA format.
2. Paste all sequences in FASTA format into a single file
In one method of performing a multiple sequence alignment, we must first create a plain text file with all the sequences we wish to align in the FASTA format. Open a word processor window on your computer (and stagger the two windows on the screen so you can easily go back and forth between them). Then paste each sequence into a single text document, with a blank line in between each.
3. Rename the sequences
For convenience (given the program we are using), in your sequence collection file, rename each sequence with the species name initials, underscore, and short name. Make sure there are no spaces in the name.
Example: rename the full FASTA description
>gi|132814447|ref|NM_00108297| Homo sapiens dopa decarboxylase (aromatic L-amino acid decarboxylase) (DDC), transcript variant 1, protein
as:
>Hsa_DDC
4. Run the alignment with Clustal Omega.
The program we will use for alignment is Clustal Omega found at the EMBL-EBI (European Molecular Biology Laboratory - European Bioinformatics Institute) website - associated with EMBO (European Molecular Biology Organization). There are others multiple sequence aligmnent programs available, including on the NCBI website.
Either paste the collected sequences into the large window, or upload the saved file (note that if uploading a file - it MUST be in a plain text/text only format).
We will use the program default settings, although we may wish to alter two output parameters (in the lower left):
Output format - try a couple different formats to see the possibilities
and
Output order [you may prefer the order in your input file to be retained - if so, change this selection to 'input' vs. the default 'aligned'
Note also that the order in which the sequences are input (that is, the order you have them in your FASTA file) can affect the alignment.
Files you can use for demonstration purposes: Example FASTA files
1. Go to Structure at NCBI
2. Search with one of the following accession numbers:
Proteins of interest related to serotonin, dopamine & biopterin synthesis
Or, try the protein of your choice, such as the protein you chose for a multiple alignment previously. Note: not all of these proteins may not have crystal structures.
3. Click on the entry, which will take you to a MMDB Structure Summary
4. View structures using FirstGlance in Jmol
Examine the structure of at least two different proteins.
5. Enter a PDB file number and wait for the image to load. Then you can view it in a variety of ways.
Extras - look at conservation of structural features of proteins. Open two windows side by side and compare structures of:
6. Human and E. coli GTP cyclohydrolases
7. Rat and C. elegans Pyruvoyl Tetrahydropterin Synthases
8. Mouse and Chlorobium (bacteria) sepiapterin reductases
9. Various aromatic amino acid hydroxylases
{kind=link}